Genes May Predict Response to Sole Sickle Cell Drug

Hydoxyurea is an almost ideal drug for many children with sickle cell anemia. It reduces the severe pain and long-term organ damage caused by sickled blood cells; it can improve lifespan; and it is cheap and fairly safe. And it’s the only FDA-approved medication for sickle cell anemia.
What’s not ideal is the variable response to the drug. “Right now, you just don’t know what’s going to happen when you start a patient on hydroxyurea,” said Nancy Green, MD, associate professor of pediatrics. “Some have only a modest response, while for others, the drug changes the disease, keeps them out of the hospital, and gets them back to their lives.”
“If the drug isn’t going to do much for a patient, we want to know so we can start thinking about other options, blood transfusions or a bone marrow transplant, that are not as easy or readily available.”
Genetic differences among patients undoubtedly play a role. With the help of a consortium she organized—with researchers from Albert Einstein, Weill-Cornell, Yale, Oakland Children’s, and the University of Rochester—Green is now starting to identify genes that eventually may help physicians better predict response to hydroxyurea.
In sickle cell disease, a mutation in the hemoglobin gene causes the hemoglobin molecules inside red blood cells to stick to each other. When that happens, the cells become damaged and lose their flexibility, causing them to assume a sickle shape. If patients also produce enough fetal hemoglobin—normally produced only in very small amounts after birth—more red blood cells retain their normal round shape and flow through the microvasculature throughout the body. Hydroxyurea works in large part because it increases the production of fetal hemoglobin, thereby reducing sickling and its complications.
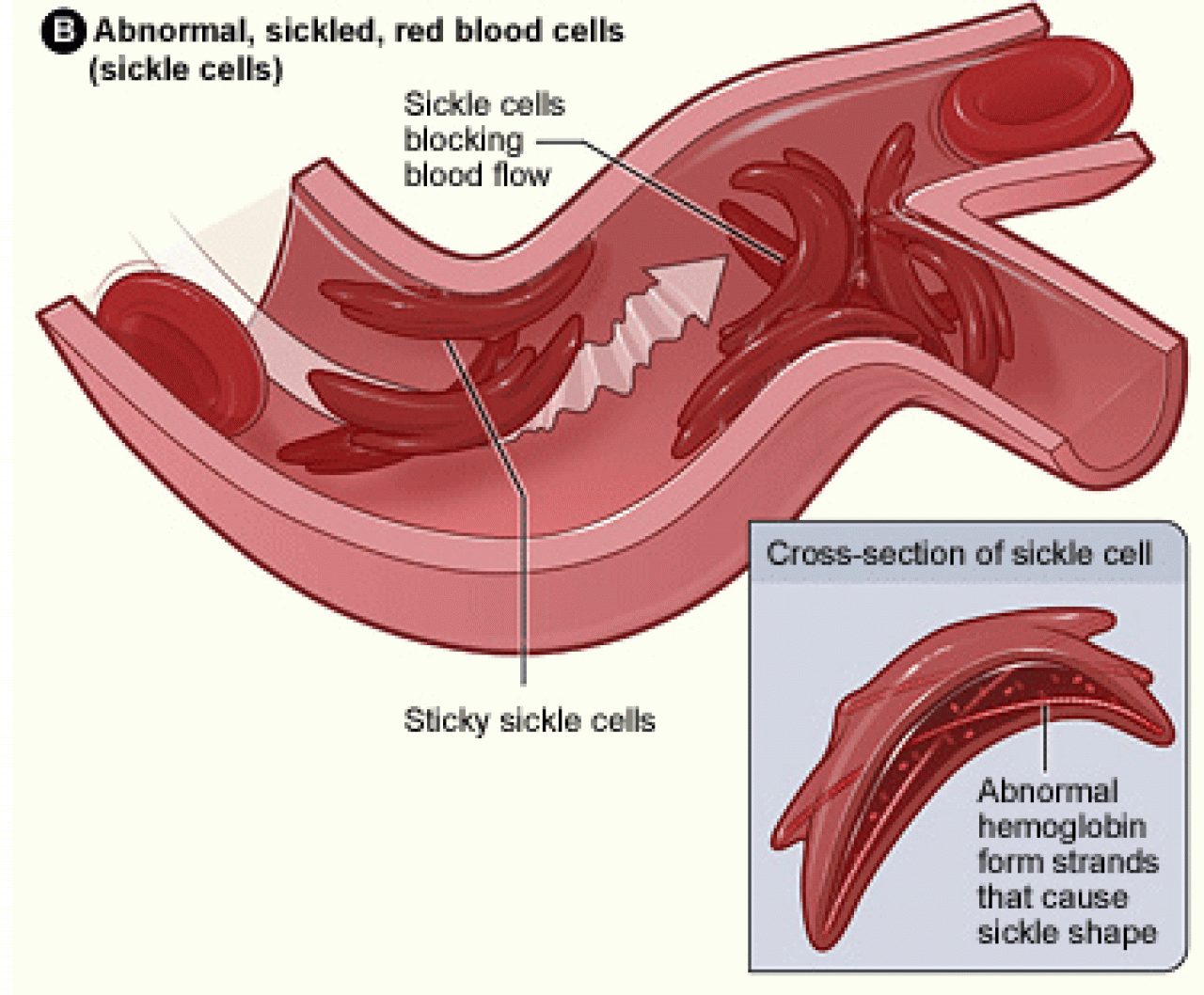
The amount of fetal hemoglobin patients can produce on their own is often an indication of the degree to which they will respond to the drug, but not always. Many patients with high baseline levels fail to mount a large fetal hemoglobin response to the drug, while fetal hemoglobin sometimes surges even in patients with low baseline levels.
A recent study published by Green and her colleagues in PloS One looked at predictors of hydroxyurea response in 117 children and found that baseline levels of fetal hemoglobin explain 33 percent of the variation in patient response.
That percentage increased to almost 50 percent when the researchers factored in each patient's type of epsilon globin gene. Green and her team also found other genes that seem to be linked to the fetal hemoglobin response to drug, but those will need to be investigated in future studies with more patients.
“Our genetic results still need to be confirmed by other researchers, and we still need to find the factors that control the other half of the variation in response, but I think we will soon get to a point when we can more reliably predict who will do well on hydroxyurea,” Green said.
More: Living with Sickle Cell, from the National Heart, Lung, and Blood Institute
“With a prediction tool, we could identify patients who should benefit but aren’t because of difficulty adhering to the daily regimen. And we could offer the drug earlier to those who are most likely to respond,” she said.
“For many children with sickle cell disease, the earlier they start using hydroxyurea, the greater impact it will have on their lives.”
CUMC researchers Katherine Ender, MD, assistant professor of clinical pediatrics, and Lorraine Clark, PhD, associate professor of clinical pathology, co-authored the study. This work was supported by NIH grants 3UL1RR024156-04S4, the St. Giles Foundation, and a gift from Henry Taub. Additional support was provided by NIH Clinical Translational Science Awards to Columbia University (UL1RR024156), Albert Einstein College of Medicine (UL1RR025750), Weill-Cornell (UL1RR024996), University of Rochester (UL1RR024160), Yale (UL1RR024139), and Oakland Children's-UCSF (UL1RR024131).